撰文:陳達慶醫師
撰稿日期:2021-08-03
遺傳性視網膜失養症 (hereditary retinal dystrophies, HRDs),也稱為基因性視網膜退化 (Inherited retinal degeneration, IRD) 是一群對視力影響相當重大的眼科疾病。這一類疾病之成因在於經遺傳或自身突變造成之基因缺陷,往往在青壯年時期或甚至更早的幼年時期就開始發病,會造成雙眼對稱性視力與視野的喪失。這類疾病大多屬於單基因疾病,可以是顯性、隱性、或性聯遺傳。根據美國 RetNet (Retinal Information Network) 之資料庫,目前已知有超過270個基因之缺陷,可造成此類疾病。依據轉譯蛋白質功能的不同,可造成視網膜感光細胞 (photoreceptors) 與視網膜色素上皮細胞 (RPE, retinal pigment epithelium) 的原發或次級退化,目前推估約影響台灣本土八千位以上的患者。

圖一、眼球結構圖:光線進入眼睛後,投射到視網膜,經由感光細胞(包含主掌中心視野和色覺辨識的錐狀細胞,以及負責暗處視之桿狀細胞),進而將這些訊號轉換成神經刺激,經視神經傳送到大腦產生影像的辨識。
(影像來源:http://91.phc.edu.tw/~tlps/good_eyes/sick/sick_index.html)
在過去,由於此類疾病牽涉的基因種類廣泛加上表現型分歧,對於臨床醫師要做病程預測、遺傳諮詢、甚或尋求治療方針,都很有難度。從臨床表徵來說,此類疾病有多種常見的表現型,是屬於「有多種可能之致病基因,不容易單從臨床表徵推估出基因診斷的」,例如最常見的視網膜色素變性 (retinitis pigmentosa),其可能的致病基因將近百個,主要症狀包含夜盲、視覺障礙、對比視力或顏色分辨能力下降、及視野狹窄等。典型的眼底表現則會在網膜上出現眾多骨針狀色素班點,並合併網膜血管縮小及視神經萎縮等症狀 (圖二A);或其他較常見的如黃斑部失養症 (macular dystrophy, 斯特格氏黃斑部失養症 Stargardt disease也包含在內) (圖二B)、錐-桿狀細胞失養症 (Cone-Rod dystrophy)、或是在幼兒時期就造成嚴重視力喪失的萊伯氏先天性黑矇症 (Leber Congenital Amaurosis) 等,皆屬於此類。也有較少部分的疾病表型,是屬於 「確認臨床診斷後,可能的致病基因較為單一或明確的」,在台灣本土來說比較常見的包括Bietti結晶樣視網膜變性 (Bietti's Crystalline Dystrophy, BCD) 主要由 CYP4V2 基因變異造成 (圖二D);性聯遺傳視網膜裂損症 (X-linked retinoschisis, XLRS) 主要由RS1 基因變異造成;以及CHM 基因變異導致的脈絡膜缺失症 (choroideremia) 等。另外值得我們注意的有兩點,一個是身體其他器官的症狀,會提供疾病診斷的線索,例如常常合併有聽力受損的尤塞氏綜合症 (Usher syndrome),其實在台灣本土的發生率也不低;另外就是在基因性視網膜退化疾病的病程晚期,許多都會造成廣泛性的視網膜萎縮,其本來獨具特色的表徵,此時反而可能會變得沒那麼明顯而可能混淆。
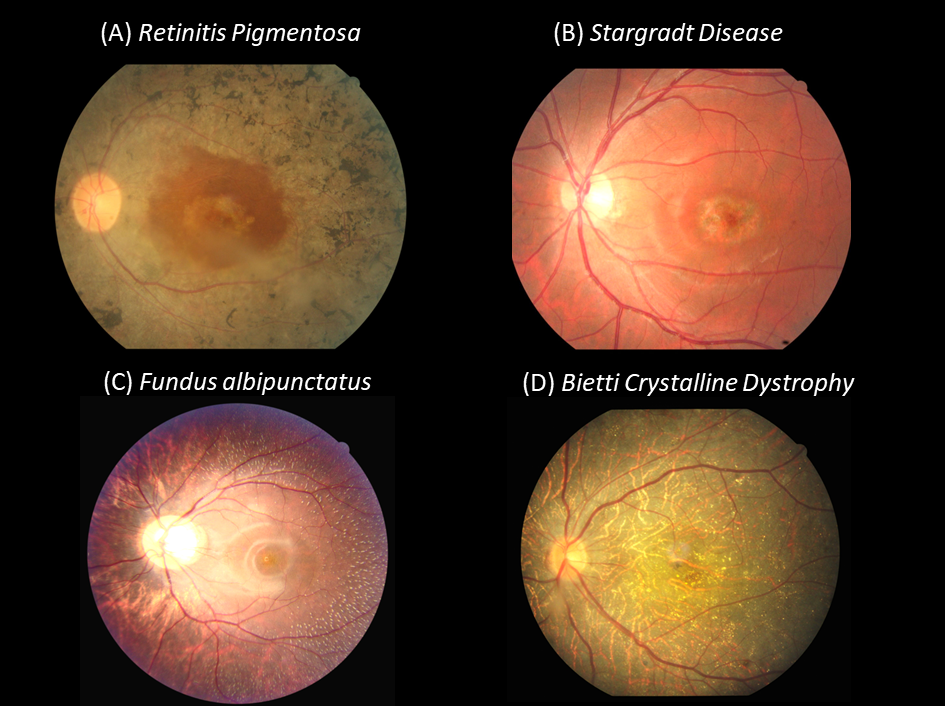
圖二
隨著近年基因醫學的進步,藉助於近年次世代定序技術 (Next-Generation Sequencing) 的發展,針對此群病友的高基因診斷率,如今已不再是夢想。為了深入了解台灣本土病患的輪廓,從2015年起,我們以台大醫院為執行單位,開啟了「台灣遺傳性視網膜退化計畫」(Taiwan Inherited retinal degeneration Project, TIP),針對台灣基因性視網膜患者與家族成員,接受全台多處醫療院所的轉診,開始進行大規模臨床診療與基因分析。近幾年來的經驗顯示,透過套組檢測作為第一線基因診斷工具,分析212個已有文獻報導過之致病基因的外顯子區域,可以有效率的達成將近六成家族的診斷率,並且注意到,有21個在台灣發生率較高的致病基因,就佔了全部病友的近一半。上述的數據從三年期312個家族的統計,到五年期475個家族的統計,趨勢非常接近,因此我們相信此結果很可能接近台灣人口族群的基因致病輪廓。[1] 有了基因資料庫與流行病學的資訊,將有助於我們了解病情的進程,以及接軌可能的新興治療。
提到近年的進展,除了基因診斷的進步,當然也不能忽略掉新興治療的開發與起步。基因治療在基因性視網膜退化領域,已經被證實是可行的途徑之一。以腺病毒相關為基因載體發展出來的基因治療,是這幾年令人振奮的好消息。其中針對 RPE65 基因缺陷所導致的遺傳性視網膜退化,如萊伯氏先天性黑矇症 (LCA) [2],已有藥物在2017年底率先完成臨床三期試驗,並通過美國 FDA 許可,成為基因療法治療單一基因缺陷疾病的全世界首例通過 FDA 驗證之藥物,可透過玻璃體切除術合併視網膜下腔注射提供病患治療。目前利用類似的藥物設計,也已有數個針對其它不同致病基因開發之藥物,陸續進入臨床試驗階段,後續發展值得期待。除了以腺病毒相關為基因載體發展出來的基因治療,更有科學家投入以基因編輯的方式 (CRISPR-based therapies),嘗試發展治療藥品,目前在前臨床期已有不錯的成果,並開始往人體臨床試驗叩關;此外透過對個別基因致病機轉的了解,針對基因缺陷的上下游蛋白質產物做為治療目標的藥物開發,或是神經保護物質的藥物開發,幹細胞治療,光遺傳學療法 (optogenetics) 或視網膜人工晶片 (Visual Prosthetics; Retinal Implant System) 等治療方式,也都持續有團隊投入嘗試與進行改良,在臨床試驗階段努力。[3-5] 希望在多個方向生醫科學的進展下,我們能逐漸對於基因性視網膜退化性疾病,有更多的了解與支持,並期望在可預期的將來,這群病友能有更好的機會接受積極性的治療,以及被治療改善的可能。
相關資訊及網頁連結:
參考文獻:
- Chen TC, Huang DS, Lin CW, Yang CH, Yang CM, Wang VY, Lin JW, Luo CL, Hu FR, Chen PL. Genetic Characteristics and Epidemiology of Inherited Retinal Degeneration in Taiwan. NPJ Genomic Medicine. 2021 Feb 19;6(1):16.
- Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017; 390:849–860.
- Zarbin M. Cell-based therapy for degenerative retinal disease. Trends Mol Med. 2016;22:115–134.
- Busskamp V, Picaud S, Sahel JA, Roska B. Optogenetic therapy for retinitis pigmentosa. Gene Ther. 2012;19:169–175.
- Dagnelie G, Christopher P, Arditi A, et al. Performance of real-world functional vision tasks by blind subjects improves after implantation with the Argus(R) II retinal prosthesis system. Clin Exp Ophthalmol. 2017;45:152–159.