撰寫人:吳婉禎醫師
撰寫日期:2020-10-02
嗜鉻細胞瘤與副神經節瘤是一種會分泌兒茶酚胺的神經內分泌腫瘤,前者源自於腎上腺髓質的嗜鉻細胞,後者源自於副交感神經節和交感神經節的嗜鉻細胞。這類腫瘤很少見,年發生率大約每百萬人口2-8例,盛行率占次發性高血壓病人的0.1-0.6%。可在任何年齡發生,最常發生在30到50歲,男、女的發生頻率相同。遺傳性嗜鉻細胞瘤與副神經節瘤通常比偶發性疾病早10年發病與被診斷。
臨床表徵
主要症狀是由於腫瘤過度分泌一種或多種兒茶酚胺(如: 正腎上腺素、腎上腺素和多巴胺)引起的。約50%嗜鉻細胞瘤患者會出現症狀,當出現症狀時,通常為陣發性。
嗜鉻細胞瘤的經典三症狀包括: 陣發性頭痛、出汗和心搏過速。持續性或陣發性高血壓是最常見的症狀,但約5-15%患者血壓正常。有症狀的患者中,約90%可出現輕度到重度且持續時間不同的頭痛,約60-70%會出汗,其他症狀包括:心悸、震顫、蒼白、呼吸困難、全身無力、恐慌發作等。而姿勢性低血壓、心肌病變、視覺模糊、體重減輕、精神疾病等症狀較少見。可能合併有碳水化合物代謝異常,如:胰島素抗性、空腹血糖上升、糖尿病。
隨著影像檢查和基因檢測的廣泛應用,無症狀的嗜鉻細胞瘤比例逐漸增加。現今約有50%的嗜鉻細胞瘤患者是由於其他原因在接受影像檢查時偶然發現腎上腺腫瘤而被診斷。
診斷與腫瘤特徵
當出現下列情況時,就要懷疑嗜鉻細胞瘤: (1) 高腎上腺素分泌症狀,(2) 頑固型或難治型高血壓,(3) 有家族性症候群,(4) 嗜鉻細胞瘤的家族史,(5) 偶然發現的腎上腺腫瘤且影像學特徵與嗜鉻細胞瘤一致,(6) 年輕性(<20歲)高血壓,(7) 特發性擴張型心肌病變,(8) 發紺型先天性心臟病。
診斷必須通過生化檢驗證實血漿或尿液中有兒茶酚胺(腎上腺素、去甲腎上腺素、多巴胺)與其代謝產物-變腎上腺素(metanephrines)、香莢杏仁酸的濃度或排泄量升高。
透過生化檢驗診斷嗜鉻細胞瘤與副神經節瘤後,才安排影像檢查確定腫瘤的位置。由於這些腫瘤約有85%位於腎上腺、95%位於腹部和骨盆腔,所以以腹部和骨盆腔的電腦斷層或磁振造影優先,會分泌兒茶酚胺的副神經節瘤的位置包括: 上腹主動脈旁(46%)、腹主動脈旁(46%)、下腹主動脈旁(29%)、膀胱(10%)、縱隔腔(10%)、頭頸部(3%)以及骨盆(2%)。另外,腎上腺髓質造影(123I-MIBG)、多巴正子掃描(18F-DOPA-PET/CT)和神經內分泌腫瘤顯影(68Ga-DOTATATE-PET/CT)也可以幫助定位。
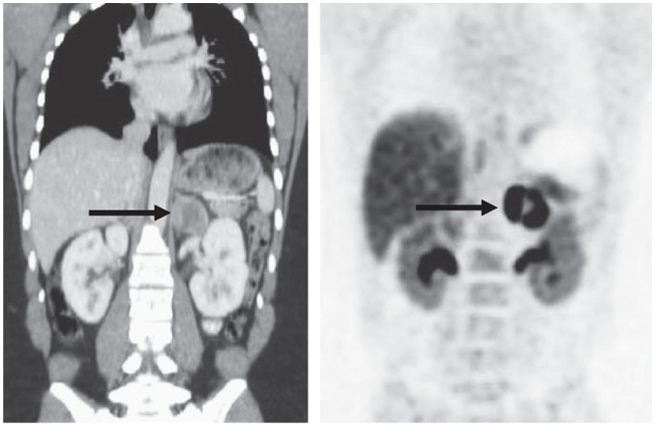
↑ 單側腎上腺嗜鉻細胞瘤(左圖)磁振造影、(右圖)多巴正子掃描(18F-DOPA-PET/CT)(圖片來源:Harrison’s Principle of Internal Medicine, 20e, Chapter 380 Pheochromocytoma)
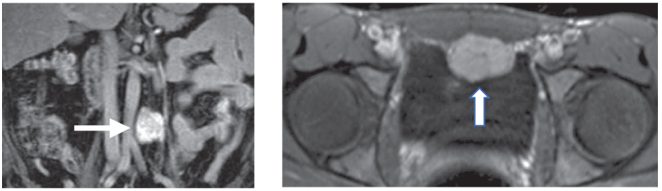
↑ 副神經節瘤(左圖)腹主動脈與大靜脈間、(右圖)骨盆腫瘤位於膀胱前壁 (圖片來源:Harrison’s Principle of Internal Medicine, 20e, Chapter 380 Pheochromocytoma)
治療、追蹤與預後
首選治療是手術完全切除,手術存活率為98-100%,取決於內分泌科醫師、內分泌外科或泌尿科醫師、和麻醉科團隊的技能。最常見的併發症是術中血壓不穩定和術後低血壓,因此術前仔細的藥物治療對於成功治療至關重要。大多數分泌兒茶酚胺的腫瘤是良性的,可以完全切除,腫瘤切除通常可以治癒高血壓。手術後大約1-2週,應追蹤24小時尿液中兒茶酚胺和與其代謝產物(後腎上腺髓素)的含量。如果數值正常表示嗜鉻細胞瘤已完整切除成功,腫瘤完全切除的良性嗜鉻細胞瘤患者的存活率幾乎與同齡同性別的一般健康族相同。如果術後24小時尿液檢出的兒茶酚胺和後腎上腺髓素的數值上升則表示有腫瘤殘存、其他病灶、或是有隱匿性轉移病灶。
雙側嗜鉻細胞瘤進行雙側腎上腺切除手術後,需要終生使用糖皮質激素和鹽皮質激素賀爾蒙補充療法。如果進行保留腎上腺皮質的雙側腎上腺切除手術,有機會保留腎上腺皮質功能,而不需要終生使用糖皮質激素和鹽皮質激素賀爾蒙補充療法。需要與內分泌科、內分泌外科或泌尿科醫師討論最適切的手術方式。
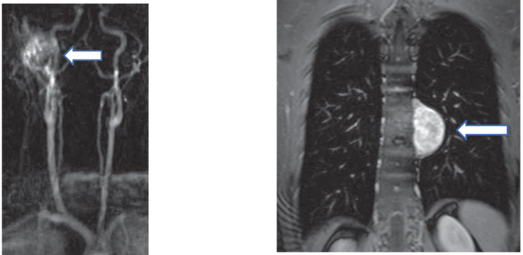
↑ 副神經節瘤(左圖)頸動脈體腫瘤、(右圖)胸腔腫瘤 (圖片來源:Harrison’s Principle of Internal Medicine, 20e, Chapter 380 Pheochromocytoma)
所有分泌兒茶酚胺的腫瘤中約有10%(8.3-13%)為惡性。轉移部位包括局部組織浸潤、骨骼,肝臟、肺臟、大網膜和淋巴結。如果可以應盡量切除轉移病灶,如果無法切除,可考慮局部治療,包括放射線治療、動脈栓塞術或射頻消融術治療等。在某些情況下,如果嗜鉻細胞瘤與副神經節瘤細胞上有生長抑素受體(SSTR),長效體抑素胜肽(octreotide)治療以及使用胜肽受體放射核種治療可能有效。
根據2017年世界衛生組織(WHO)的資料,所有嗜鉻細胞瘤與副神經節瘤都有一定的轉移潛力。在長期隨訪中,良性嗜鉻細胞瘤或副神經節瘤發生復發性或轉移性疾病的風險約為15%,有些甚至可能在50年後才發生。因此,所有分泌兒茶酚胺的嗜鉻細胞瘤與副神經節瘤的患者都需要終生定期追蹤! 每年應進行生化檢查包括: 血漿游離變腎上腺素的濃度以及24小時尿液中兒茶酚胺、香莢杏仁酸、變腎上腺素的排泄總量來評估是否有復發性或轉移性疾病。當血漿或尿液中有兒茶酚胺與後腎上腺髓素的濃度升高時,才需要進行後續電腦斷層或磁振造影檢查。
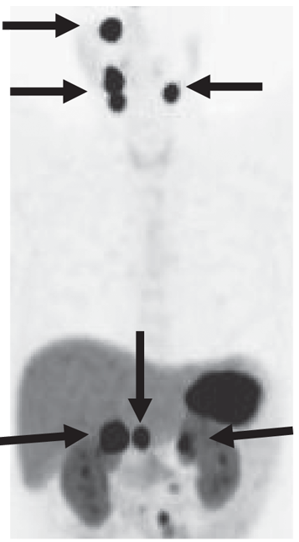
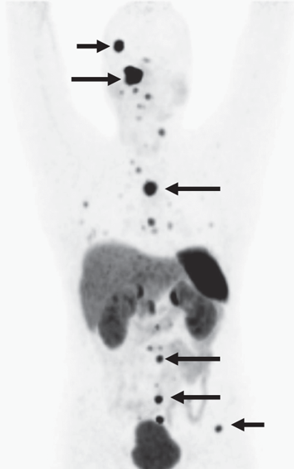
↑ 多發性與轉移性嗜鉻細胞瘤與副神經節瘤(左圖)副神經節瘤綜合症、(右圖)轉移性嗜鉻細胞瘤
(圖片來源:Harrison’s Principle of Internal Medicine, 20e, Chapter 380 Pheochromocytoma)
家族性與遺傳性
大多數分泌兒茶酚胺的腫瘤是偶發性的(sporadic),但約有40%的患者是家族性疾病的一部分,這些患者較容易出現雙側腎上腺嗜鉻細胞瘤或副神經節瘤。遺傳性腫瘤的患者通常比偶發性腫瘤較年輕發病以及診斷。而源自於副交感神經節的副神經節瘤中有約25%是遺傳綜合症的一部分。
有幾種與腎上腺嗜鉻細胞瘤有關的家族性疾病,它們均是顯性遺傳,包括:林道症候群(von Hippel-Lindau, VHL)、第二型多發性內分泌瘤(multiple endocrine neoplasia type 2, MEN2)、以及較少見的第一型神經纖維瘤(neurofibromatosis type 1, NF1)。在這些疾病中,嗜鉻細胞瘤的發生頻率在VHL中為10-20%、在MEN2中為50%、在NF1中為0.1-5.7%。
目前已發現20多個基因的體細胞突變或生殖細胞突變和嗜鉻細胞瘤與副神經節瘤有關。惡性腫瘤在MEN2或VHL患者中很少見,但由SHDB突變引起的家族性副神經節瘤患者中惡性腫瘤或轉移性疾病的機率高。在家族性疾病中,大的腫瘤(>5公分)或副神經節瘤的患者,復發率較高。

↑ 綜合症與其相關基因
由於嗜鉻細胞瘤與副神經節瘤(1)存在致命性陣發性疾病的風險,(2)通過手術切除腫瘤可以治癒相關的高血壓;(3)至少10%是惡性的;以及(4)有40%是家族性的,在發病者中如有偵測到遺傳性基因突變,應讓其他家庭成員及早接受基因諮詢與檢測。因此及早懷疑、早期診斷與接受治療至關重要。
參考資料
- Williams Textbook of Endocrinology, 14e, Chapter 16 Endocrine Hypertension by William F. Young Jr.
- Harrison’s Principle of Internal Medicine, 20e, Chapter 380 Pheochromocytoma by Hartmut P.H. Neumann.
- Jacques W. M. Lenders, Quan-Yang Duh, Graeme Eisenhofer, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014; 99: 1915-42.
- Herbert Chen, Rebecca S. Sippel, M. Sue O’Dorisio, et al. The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors - Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer. Pancreas 2010; 39: 775-83.
- Farrugia FA, Martikos G, Tzanetis P, et al. Pheochromocytoma, diagnosis and treatment: Review of the literature. Endocrine Regulations 2017; 51: 168-81.
- Andrew S Davison, Danielle M Jones1, Stuart Ruthven, et al. Clinical evaluation and treatment of phaeochromocytoma. Annals of Clinical Biochemistry 2018; 55: 34-38.
- Crona J, Taïeb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocrine Reviews 2017; 38: 489-515.